Select Taxa and Orthologs to be Included in a Phylogenomic Dataset
To select taxa and orthologs to be included in the final phylogenomic dataset users can run the scripts select_taxa.py and select_orthologs.py respectively. Each will generate a .tsv file (contents explained below) that will serve to select either taxa or orthologs to include and both .tsv files are used as input for the script prep_final_dataset.py downstream.
NOTE: If all taxa and all orthologs in the database are to be included in the final phylogenomic dataset steps 1 and 2 are not necessary. Skip directly to step 3.
-
Select taxa for the final phylogenomic analyses:
select_taxa.py[OPTIONS]Optional arguments:
-
--to_exclude taxa_to_exclude.txtPath to a text file of groups or individual taxa to exclude from the final phylogenomic matrix -
--to_include taxa_to_include.txtPath to a text file that will re-include groups or individual taxa -
--chimeras chimeras.tsvPath to a .tsv file that contains a Unique ID, higher and lower taxonomic designations of the chimera, and the Unique IDs of the taxa in the database to collapse, for each chimera one per line (Table 4) -
-h,--helpShow this help message and exit
Default
select_taxa.pyoutput:-
a .tsv file
select_taxa.tsvwith five columns:-
Unique ID
-
Higher Taxonomy
-
Lower Taxonomy
-
Completeness (percentage of orthologs) from the database present in the taxon
-
Include in Subset
-
-
a plot showing completeness (number of orthologs) in each taxon. Groups of taxa are colored by level of completeness in increments of 10% (Figure 5).
NOTE: Detailed explanation of to the
--to_excludeand--to_includeoptions and their input files:By default all taxa will be included. These options were designed to decrease the amount of manual manipulation of
select_taxa.tsvgenerated in this first run ofselect_taxa.py. Both options take a file created by the user that has one column with an organism’s Unique ID or a taxonomic rank (higher or lower) from the metadata to be excluded or included in downstream steps. The options can be used individually or in conjunction with one another to implement decisions on taxa selection in an automated fashion. For example, if all taxa in the eukaryotic assemblage “Amoebozoa” are to be excluded from downstream phylogenomic analyses provide the--exclude_taxaoption with a file (named anything) that contains “Amoebozoa” as the first entry of the column. This will result in all amoebozoans being marked as “no” in the “Include in Subset” column ofselect_taxa.tsvwhen it is generated. If a user wanted all amoebozoans excluded except Dictyostelium discoideum (Unique ID = Dictdisc) provide the--exclude_taxaoption with a file (named anything) that contains “Amoebozoa” as the first entry of the column as before and the--to_includeoption with a file (named anything) with “Dictdisc” as the first entry of the column. This will result in all amoebozoans being marked as “no” in the “Include in Subset” column ofselect_taxa.tsvexcept D. discoideum which will be marked “yes.”NOTE:
--to_includeis not necessary it only serves to ease the burden of manual taxa selection in cases as outlined above. All taxa are marked to include by default so it is NOT necessary to provide--to_includewith a list of all taxa you wish to include in downstream steps.NOTE: Decisions provided to
--to_includewill override decisions provided to--to_excludeso be cautious.Alternatively, all changes can be made manually by opening
select_taxa.tsvand changing taxa designations from “yes” to “no”.BrevCHIM Obazoa Breviatea Breviate spp. Brevanat Lenilimo Pygsbifo Table 4: Example input .tsv file for the
--chimeraoption ofselect_taxa.py. Here a chimera will be made from three breviates in the database. The chimera will have the Unique ID ”BrevCHIM”, the higher taxonomy ”Obazoa”, the lower taxonomy ”Breviatea” and will be made from Brevanat (Breviata anathema), Lenilimo (Lenisia limosa), Pygsbifo (Pygsuia biforma).
Figure 5: Plot produced by select_taxa.py. Taxa Unique IDs are shown at the bottom of the figure across the x-axis. The percentage of total orthologs present in a taxon (completeness) is shown on the y-axis. The total number of taxa is shown across the top of the figure on the x-axis. The bars are colored by level of completeness in increments of 10%. Legend explaining color coding is shown in the top right of the figure -
-
Select orthologs to be included in the final phylogenomic dataset
select_orthologs.py [OPTIONS]Optional arguments:
-
--out_group out_group.txtPath to text file containing out group taxa Unique IDs -
--chimeras chimeras.tsvA .tsv containing chimeras and taxa to collapse -
-n,--gene_number <N>Number of genes for analysis -
-c,--percent_complete <N>Threshold for percent completeness -
-h,--helpShow this help message and exit
select_orthologs.pyoutput:-
a tsv file
select_orthologs.tsvwith three either three (default) or five columns:-
Ortholog - ortholog name
-
Total Completeness - percentage of all taxa to be included in the final phylogenomic dataset the ortholog is present in.
-
In-Group Completeness (only present if
--out_groupoption is utilized) -percentage of In-group taxa only to be included in the final phylogenomic dataset the ortholog is present in. -
Out-Group Completeness (only present if
--out_groupoption is utilized) - percentage of Out-Group taxa only to be included in the final phylogenomic dataset the ortholog is present in. -
Include in Subset
-
-
a plot showing completeness (number of taxa) of each gene. Groups of genes are colored by level of completeness in increments of 10% (Figure 6).
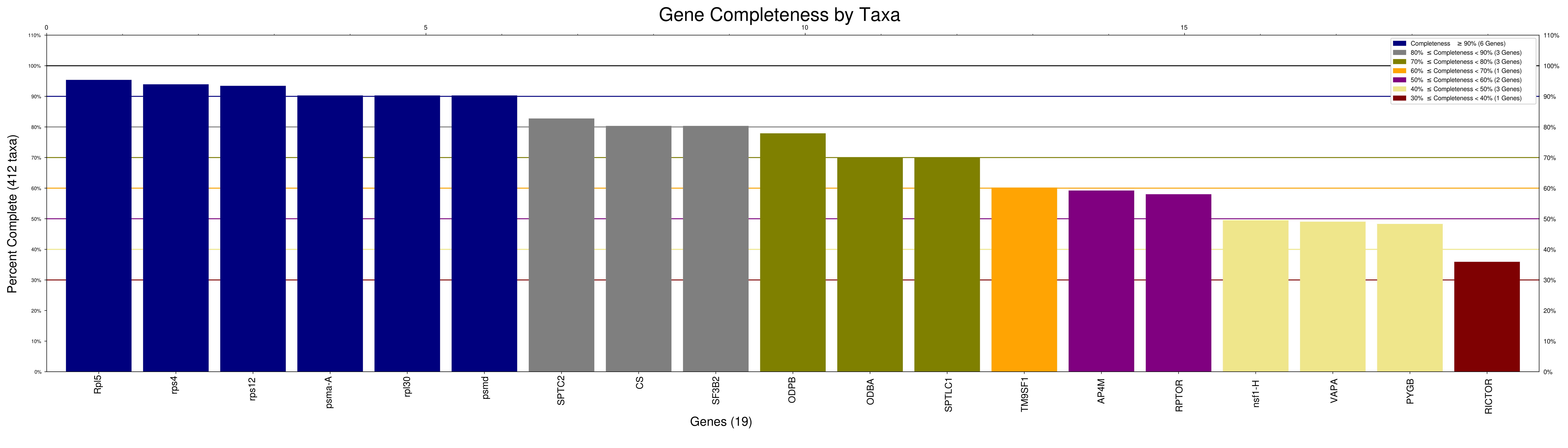
Figure 6: Plot produced by select_orthologs.py. Ortholog names are shown at the bottom of the figure across the x-axis. The percentage of total taxa present in an ortholog (completeness) is shown on the y-axis. The total number of genes is shown across the top of the figure on the x-axis. The bars are colored by level of completeness in increments of 10%. Legend explaining color coding is shown in the top right of the figure. -
-
Collect orthologs and taxa to be included in the final phylogenomic dataset
prep_final_dataset.py [OPTIONS]Optional arguments:
-
-o,--output <out_dir>Path to user-defined output directory- default
./prep_final_dataset_<M.D.Y>
- default
-
-h,--helpShow this help message and exit
Default
prep_final_dataset.pyoutput:-
a directory
prep_final_dataset_<M.D.Y>that contains:- a fasta file of each ortholog selected to be in the final phylogenomic dataset each containing only taxa selected to be in the final phylogenomic dataset.
-