PhyloFisher Package Contents
- Manually curated database of 240 orthologs and their paralogs from 304 eukaryotic taxa. These taxa cover the known diversity of eukaryotes.
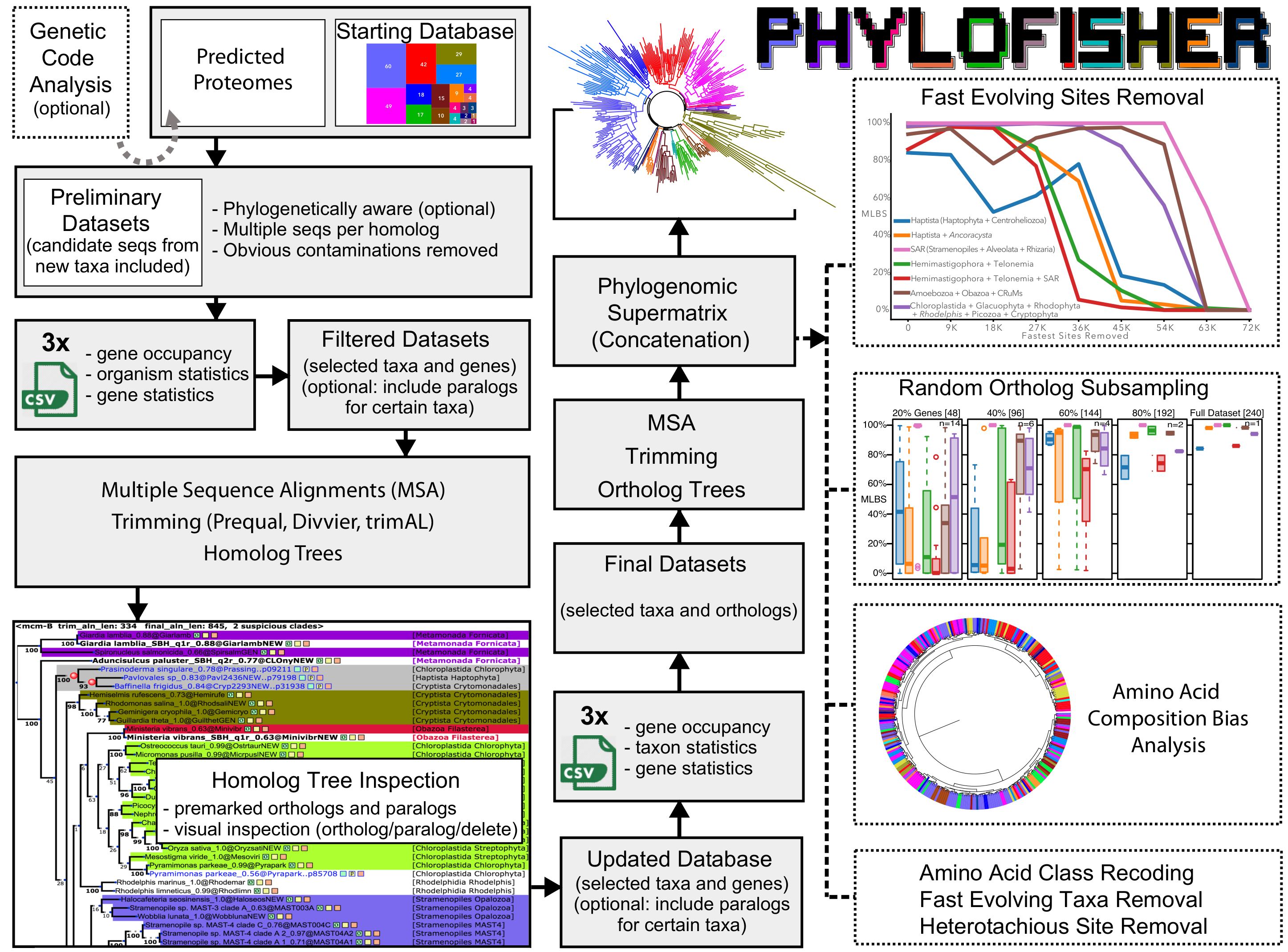
- Main Workflow tools for:
- The preparation of custom databases composed of protein coding genes in lieu of the provided database
- Ortholog and paralog “mining” from input proteomes
- Evaluating completeness in the database of newly input data
- Automated removal of non-homologous sites, alignment, trimming, length filtering and single gene tree construction
- Visualization of single gene trees and interactive selection of orthologs, paralogs, and removal of contamination
- Generation of final statistics post ortholog selection and contamination removal
- Can be subset by user provided thresholds of completeness or by assigned taxonomy
- Creation of chimeric taxa for use in phylogenomic analyses
- Automated concatenation of phylogenomic matrices
- Utilities for:
- Alternative genetic code prediction
- Compositional bias testing (amino acid re-coding)
- The removal of fast evolving sites from a phylogenomic dataset
- The removal of fast evolving taxa from a phylogenomic dataset
- Detection and removal of the most heterotachious sites in a phylogenomic dataset
- Examining the number of occurrences of clades of interest in bootstrap trees
- Matrix construction from randomly re-sampled genes from a phylogenomic dataset
- Binning single-protein trees based on Relative Tree Certainty (RTC)
- Collapsing of multiple proteomes to produce a single “most complete” proteome with regards to the database (useful for single-cell data and to form chimeric taxa from closely related species)