Ortholog Selection via the Fisher Algorithm
Ortholog selection and paralog demarcation is performed using the python script fisher.py (Figure 3) that requires two inputs: predicted proteome(s) to be added to the database and an input-metadata file containing key information about them. To remove redundancy in the input proteome (typical when proteomes are predicted in silico from transcriptomic data), fisher.py invokes CD-HIT (Fu et al., 2012) with the option -c 0.98, which clusters sequences globally at a similarity threshold of 98%. Next, hmmsearch from the software package HMMER1 (Mistry et al., 2013) is run on the input proteome using a pre-computed profile Hidden Markov Model (HMM) of each protein alignment in the database. Up to a user-defined number of sequences are collected (default = 5) that meet the significance threshold (e-value < 1e -10). If no sequence meets the significance threshold, the script moves on to the next protein. If sequences are found (up to the user-defined number), they are prioritized based on the level of significance (i.e., the sequence with the most significant hit is preliminarily denoted as the most likely putative ortholog with other included sequences preliminarily denoted as putative paralogs). From here, the algorithm proceeds in either of two directions, depending on information given in the input metadata file. In the two routes outlined below, sequences can be added to or removed from the initial sequences collected by hmmsearch, as they meet or fail to meet the outlined criteria. The prioritization of a sequence can also change within the list based on the criteria outlined below. At the end of either route, if only one sequence remains, it is considered the putative ortholog for the input taxon. If more than one remains, the sequence with the highest priority in the list is considered the putative ortholog and the other retained sequences are considered putative paralogs. All retained sequences are added to the corresponding single-protein alignment.
Default Route
If no related species present in the database is/are listed in the “Blast Seed” column of the input metadata file to use as specific queries, each sequence collected from the initial hmmsearch is used as the query in a search using DIAMOND(Buchfink et al., 2015) against OrthoMCL (Chen et al., 2006) with the options blastp -e 1e-10 –more-sensitive. Any sequence that has a significant hit (e-value < 1e -10) to a bacterial orthogroup in OrthoMCL is discarded along with any sequence that does not have a significant hit to the OrthoMCL orthogroup corresponding to the respective profile HMM used to retrieve the sequence. Retained sequences are again used as queries in a DIAMOND search using the parameters given above against the database. If the query sequence’s best blast hit is a sequence corresponding to the profile HMM that retrieved the query sequence in the initial search, the sequence is retained. If the sequence’s best blast hit is a sequence from another alignment, the sequence is still retained, but is written out to the file non_corresponding_hits.txt with a note on which protein from the database represents the best blast hit. The remaining sequence that has the highest priority based on the initial hmmsearch is considered to be the putative ortholog and any other surviving sequences are labelled putative paralogs.
Phylogenetically Informed Route
The input metadata file gives users the option to specify taxa already in the database whose sequences will be used as queries for blast searches against the new organism’s proteome; typically these should be closely related to the newly added taxon. Any number of species already present in the database may be chosen to serve as specific queries, but the algorithm will use them in the order provided, and, in cases outlined below, may not proceed to subsequent taxa. If species to be used as specific queries are listed in the input metadata file, fisher.py will initially pick the sequence representing a particular orthologous gene group (“ortholog” for simplicity) from the first organism listed, if present. If absent in the database, fisher.py will sequentially check for the ortholog from the remaining taxa listed. If the orthologs from one or more of the subsequent taxa are found, the algorithm proceeds as outlined below. If no ortholog from all listed organisms can be found, fisher.py will proceed to the default route for this particular protein. If an ortholog is present in at least one of the listed species, then its sequence is used as the query in a BLAST (Camacho et al. 2009) search against the input organism’s predicted proteome. If no significant hit (e-value < 1e -10) is found, fisher.py will use the ortholog of the next listed species, if one is provided. If no other species is listed, the ortholog is not present in the database for the listed species, or if the orthologs of all listed species do not return a significant BLAST hit, the protein is skipped for the input taxon. If significant BLAST hits are found, up to a user-defined number of those (default = 5) are collected and examined further. The sequences from the original hmmsearch are reprioritized based on the level of significance of the BLAST hit. The sequence with the most significant hit becomes the priority sequence, unless it was not also collected by the initial hmmsearch: any sequence that produces a significant BLAST hit but was not collected in the initial hmmsearch is discarded. The retained sequences are then used as queries against OrthoMCL. Any sequence that has a significant hit (e-value < 1e -10) to a bacterial orthogroup in OrthoMCL is discarded along with any sequence that does not have a significant hit to the OrthoMCL orthogroup corresponding to the respective profile HMM used to retrieve the sequence. Next, all the sequences that have passed the filtering in the previous step are compared with blast against the database of conserved marker proteins. If the query sequence’s best blast hit is a sequence corresponding to the profile HMM that retrieved the query sequence in the initial search, the sequence is retained. If the sequence’s best blast hit is a sequence from another alignment, the sequence is still retained, but is written out to the file non_corresponding_hits.txt with a note on which protein from the database represents the best blast hit. Sequences are then added to the corresponding dataset from the database and aligned using MAFFT (Katoh and Standley, 2013) with the parameters –auto –reorder, trimmed with trimAl (Capella-Gutiérrez et al. 2009) with a gap threshold of 0.2, and subjected to phylogenetic tree reconstruction via FastTree (Price et al., 2010) with default parameters. The resulting tree is examined using the python package ETE3 (Huerta-Cepas et al., 2016). By default sequences that branch sister to or within a clade composed of organisms with the same assigned higher taxonomy are retained while sequences that branch outside of the assigned higher taxonomy are eliminated unless no sequence branches with sequences assigned the correct higher taxonomy. This happens regardless of previous criteria. Higher taxonomy for taxa is derived either from the database’s metadata file or the input metadata file. Finally, a length filtration step will remove any sequences that have more than 70% gaps in the trimmed alignment. All retained sequences are then added to their corresponding alignments from the database, with the sequence that received the highest level of priority being denoted the putative ortholog and all others denoted as putative paralogs.
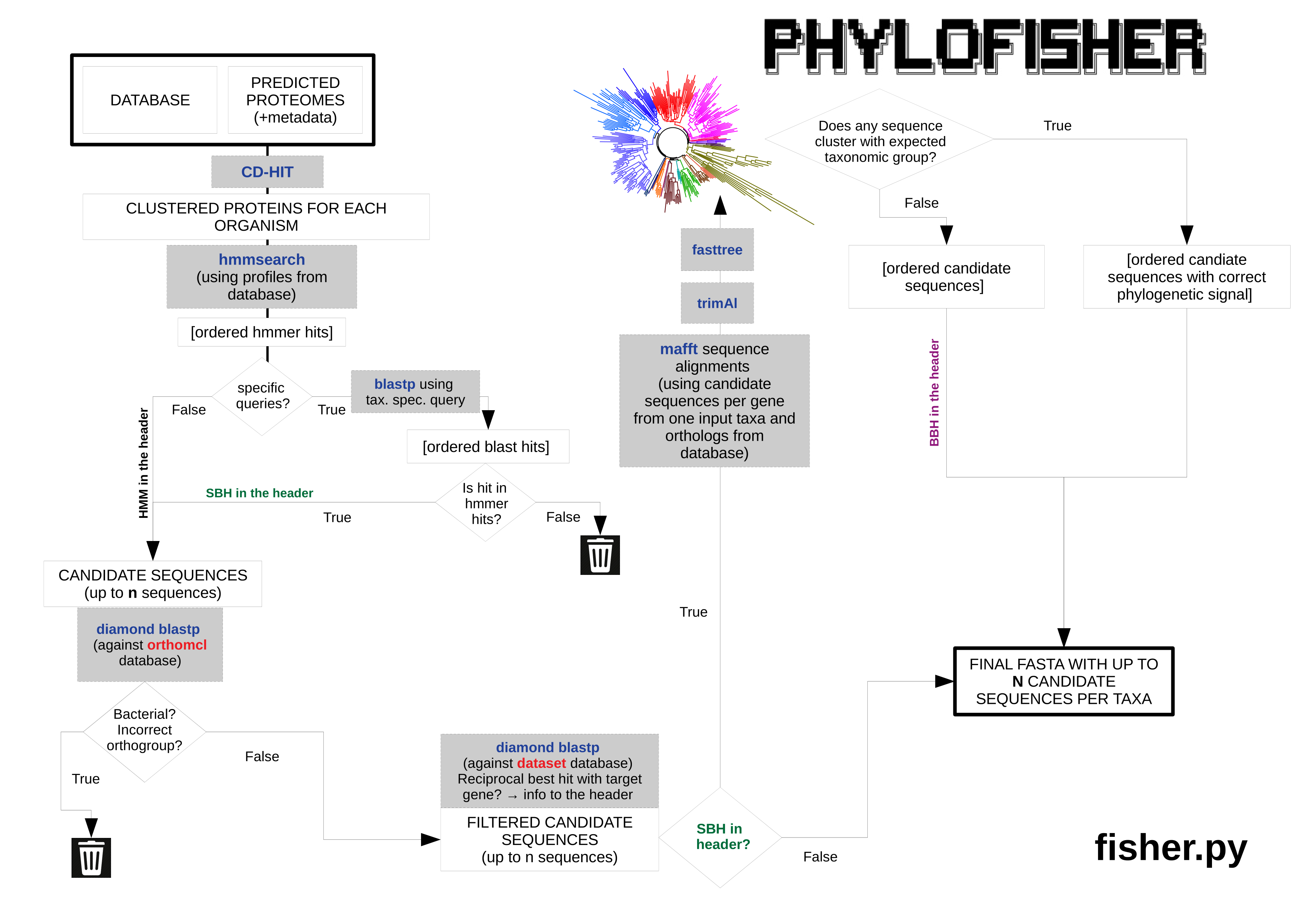
fisher.py. Briefly, each predicted proteome of a new taxon to be added is processed through either a default route, or a phylogenetically aware route that utilizes the manually curated orthologs from closely related taxa chosen by the user (and present in the database) as search queries against the proteome of the new taxon. Up to a user-defined number of collected sequences are reprioritized or eliminated based on a set of criteria designed to maximize correct demarcation of the desired ortholog and related paralogs while avoiding contaminating sequences. See section “Ortholog Selection via the Fisher Algorithm” of this guide for a detailed description of the logic, third-party software and associated parameters utilized.